

精神神経疾患の確定診断は,今日においても神経病理学的診断により行われている.アルツハイマー病,ピック病,進行性核上性麻痺,大脳皮質基底核変性症,嗜銀顆粒性認知症,パーキンソン病およびレビー小体型認知症,多系統萎縮症,前頭側頭葉変性症や筋萎縮性側索硬化症などは,神経病理学的に疾患特異的な封入体とそこに凝集蓄積する特異的蛋白によって分類されている.神経変性疾患は,神経細胞内に生理的に存在する可溶性蛋白が,さまざまな修飾を受けて不溶化し,細胞内に凝集蓄積して病理学的に封入体を形成する蛋白症(蛋白変性症)として認識されている.大脳皮質の巣症状のみならず,幻覚・妄想などの精神病様状態,気分障害,行動・心理症状なども病理学的背景をもつことに留意が必要である.正確な臨床情報に基づく病理学的に確定診断された症例の蓄積が,蛋白変性過程の病態解明や創薬などの治療法開発につながる.
精神科臨床に役だつ神経病理学の知識
愛知医科大学加齢医科学研究所
精神神経学雑誌
120:
290-301, 2018
<索引用語:神経病理, 封入体, 蛋白症, 前頭側頭葉変性症, TDP-43>
はじめに
精神神経疾患の確定診断は,今日においても神経病理学的診断により行われている.神経変性疾患が器質的な病変を示すことは自明なことであるが,近年の神経病理学的解析の蓄積により,幻覚・妄想などの精神病様状態,気分障害,行動・心理症状(behavioral and psychological symptoms of dementia:BPSD)なども,脳の器質的病変により生じうることが認識されるようになってきている.神経病理学的診断は変性(神経細胞脱落の分布)と神経細胞やグリア細胞内に観察される異常な構造物である封入体の形態,および封入体を構成する蛋白に基づいて評価される.本稿では,精神科で遭遇する機会の多い疾患を中心に,精神科臨床に役だつ神経病理学の知識を概説する.
I.蛋白症としての神経変性疾患
神経変性疾患は,潜在的に発症し,緩徐進行性で原因不明な疾患群を総称しているが,これらの疾患は細胞内に生理的に存在する可溶性蛋白が,さまざまな修飾を受けて不溶化し,細胞内に凝集蓄積して病理学的に封入体を形成する蛋白症(蛋白変性症)として認識され,分類されている.アルツハイマー病(Alzheimer's disease:AD)やピック病(Pick's disease:PiD),進行性核上性麻痺(progressive supranuclear palsy:PSP),大脳皮質基底核変性症(corticobasal degeneration:CBD),嗜銀顆粒性認知症(dementia with grains:DG),パーキンソン病(Parkinson's disease:PD)およびレビー小体型認知症(dementia with Lewy bodies:DLB),多系統萎縮症(multiple system atrophy:MSA),前頭側頭葉変性症(frontotemporal lobar degeneration:FTLD),筋萎縮性側索硬化症(amyotrophic lateral sclerosis:ALS)などは,神経病理学的に疾患特異的な封入体とそこに凝集蓄積する特異的蛋白によって分類されている(表).各疾患の神経病理学的特徴を以下に述べる.
1.アルツハイマー病(AD)
ADの病理診断は,老人斑(senile plaques:SP)とアルツハイマー神経原線維変化(AD-neurofibrillary tangles:AD-NFT)が生理的老化による出現量と分布を超えて,大量かつ広範囲に蓄積することを基本とする.SPは細胞外にβアミロイド蛋白(amyloid β-protein:Aβ)が凝集蓄積した封入体であり,AD-NFTは神経細胞内にタウ蛋白が凝集して束状の線維性構造を示す封入体である(表,図1).
ADでは,ある程度進行すると大脳皮質のびまん性萎縮,海馬領域の萎縮や神経細胞脱落が明瞭となる(図1).Aβは,1-42のペプチドからなるAβ(1-42)と血管壁への沈着が強いAβ(1-40)があり,SPでは両者が沈着する.アミロイドアンギオパチー(cerebral amyloid angiopathy:CAA)ではAβ(1-40)の沈着が優位となるが,毛細血管に沈着する場合にはAβ(1-42)が優位となる.SPは,初期には後頭葉や前頭葉下面,側頭葉外側から沈着し始め,大脳新皮質から辺縁系,基底核・視床,脳幹部・小脳へと広がることが報告されている5)31).2012年のADの病理診断基準20)では,SPの病理学的評価法は,変性神経突起を伴うSPの密度によるCERAD分類18),大脳皮質への広がりによるBraak分類5),大脳から基底核,視床,脳幹部,小脳への広がりを評価するThal分類31)の3つの方式が採用されている.
タウは物質輸送にかかわる微小管の重合促進と安定化に作用する蛋白で,第17番染色体長腕17q21.2に存在するタウ遺伝子(microtubule associated protein tau:MAPT)の選択的スプライシングにより,6種類のアイソフォームが形成される.微小管との結合にかかわる繰り返し配列が4つの4リピートアイソフォーム(4R)と,3つの3リピートアイソフォーム(3R)に大別される.AD-NFTは6種類すべてのタウアイソフォームから構成され,超微形態的にpaired helical filament(PHF)と呼ばれる線維構造をとり,リン酸化やユビキチン化などの修飾を受けて蓄積している.
Braak, H. らは多数の剖検脳の詳細な検討により,AD-NFTの出現分布を6段階に分類した5)6).初期には嗅内野と経嗅内野の境界領域に出現(stageI/II)し,徐々に後頭側頭回(stage III)から等皮質(stage IV/V)へ広がり,最も高度になると一次視覚野や運動野(stage VI)に広がることを示した.臨床的な認知症は,AD-NFTの出現量によく合致することが知られている.
Murray, M. E. らはAD-NFTの病変分布に3つの亜型があることを報告した21).大脳皮質にびまん性にNFTが出現する典型的AD型(75%)以外に,辺縁系優位型(14%),hippocampal sparing型(11%)に分類され,hippocampal sparing型では大脳皮質のAD-NFTの出現が多く,海馬領域の出現がより少なく,海馬の萎縮が軽いことが報告されている.自験例においても,CBDと臨床診断されたADはhippocampal sparing型に相当していた.
2.タウオパチー
病理学的に神経細胞内やグリア細胞内にタウ蛋白が蓄積する疾患を孤発性,遺伝性を含めてタウオパチーと総称する.前述のAD-NFTは3Rタウと4Rタウの両者から構成されているのに対して,PiDのピック球は3Rタウが構成成分であり,PSPやCBD,DGでは4Rタウが神経細胞やグリア細胞に蓄積しており,疾患ごとに封入体を構成するタウ蛋白のアイソフォームの特異性に相違がある(表).
1)ピック病(PiD)
PiDは主として初老期に発症し緩徐に進行する人格変化や行動異常,言語機能障害を特徴とし,病理学的には前頭葉と側頭葉の前方領域優位に葉性萎縮と呼ばれる限局性の萎縮を認める.萎縮の高度な部位では脳回はナイフの刃様萎縮(knife-edge atrophy)を示す(図2).しばしば左右差を伴い後方の上側頭回や頭頂後頭葉などでは変化が軽いことが多い.組織学的には皮質表層優位の神経細胞脱落とグリオーシス,残存する神経細胞内に3Rタウから構成される好塩基性の類円形封入体であるピック球を認める(図2).
PiDは,今日のFTLDの原型となった疾患であり,大脳のびまん性萎縮を示すADと対比されるが,愛知医科大学加齢医科学研究所の約5,500例の剖検例中,ADは約200例であるのに対してPiDは6例であり,その頻度はADに比して極めて低い.初老期発症を特徴とするものの,病理学的に確定診断された自験例の発症年齢は平均69歳(59~77歳)であり,高齢者の認知症にも含まれている点で留意が必要である.PiDの疾患概念は,ピック球の有無を問わなかったために,混乱をきたしたが,「ピック球のないPiD」から今日のTDP-43蛋白変性を伴うFTLDが分離された経緯は,変性疾患における封入体と特異的構成蛋白の重要性を決定的なものにしたと言っても過言ではない.
2)進行性核上性麻痺(PSP)と大脳皮質基底核変性症(CBD)
PSPとCBDは,神経細胞とグリア細胞に4 Rタウが蓄積する疾患で,両疾患はともにグリアのタウ蓄積が優位であることを特徴とし,PSPではtufted astrocyte,CBDではastrocytic plaqueというアストロサイトの封入体が病理診断指標となっている11)(図3, 図4).PSPの臨床像は,Steele, J. C. らによれば易転倒性,核上性眼球運動障害,頸部後屈が強調されてきたが,その後の剖検例の蓄積により臨床像はより多彩であることが明かにされている29).2017年のPSPの新しい臨床診断基準では,眼球運動障害,姿勢安定性,無動,認知機能障害を主要4症候として,それらの組み合わせにより臨床病型を8型にPSP-RS(PSP-Richardson-syndrome),PSP-OM(PSP with initial predominance of ocular motor dysfunction predominant),PSP-PI(PSP with initial predominance of postural instability),PSP-P(PSP with Parkinsonism resembling idiopathic Parkinson's disease),PSP-F(PSP with frontal lobe cognitive or behavioral presentations),PSP-PGF(PSP with progressive gait freezing),PSP-CBS(PSP with corticobasal syndrome),and PSP-SL〔PSP with nonfluent/agrammatic primary progressive aphasia(nfaPPA)and progressive apraxia of speech(AOS)〕に分類している9).
PSPの病変分布は,淡蒼球・視床下核,黒質,脳幹部被蓋,小脳歯状核の変性が中核であり,それに加えてさまざまな程度に中心前回を含む前頭葉穹隆面の変性を伴っている(図3).臨床像の多様性は,神経細胞脱落とタウの蓄積による病変分布のアクセントに由来している33).
Rebeiz, J. J. らによれば,他人の手徴候や,片側性の肢節運動失行などの大脳皮質徴候が特徴的とされたが,その後の剖検例の蓄積により臨床病理像のスペクトラムが広いこと,臨床的にCBDと診断された症例の病理像には,CBD以外の疾患が半数近く含まれることから,臨床診断には大脳皮質基底核症候群(corticobasal syndrome:CBS)を用い,病理診断にCBDを用いることが推奨されている25).2013年にCBDの主要な臨床病型が提唱され2),CBD-CBS(CBD with corticobasal syndrome),CBD-FBS(CBD with frontal behavioral-spacial syndrome),CBD-naPPA(CBD with nonfluent-aggrammatic variant of primary progressive aphasia),CBD-PSPS(CBD with PSP syndrome)の4つに大別された.
CBDの病変分布は,大脳皮質・白質,淡蒼球,黒質,視床下核などを中心とするが,大脳皮質の左右差や病変分布とその程度,脳幹部や小脳歯状核などの変性程度には症例ごとの差異があり,臨床像の多様性の背景となっている(図4)33).
PSPとCBDは,大脳皮質,淡蒼球,視床下核,黒質などに共通した病変分布をもち,臨床像が類似するため,しばしば臨床診断と病理診断の逆転が起こる.つまり臨床診断はCBDだったが,病理診断はPSPであったということが生じる.これはPSPとCBDの病変分布の共通性に起因しているためであり,確定診断には病理学的検索が必須である.
3)嗜銀顆粒性認知症(DG)
1987年,Braakらは側頭葉の内側面の灰白質にGallyas-Braak嗜銀染色で染色される嗜銀性顆粒状封入体(argyrophilic grain:AG)を形成する認知症をDGと呼称することを提唱した3)4).AGは高齢者の扁桃核,迂回回,海馬傍回などの内側側頭葉に高頻度に出現する4Rタウの封入体である(図5).しばしば肉眼的に側頭葉内側面に左右差を伴い,ことに迂回回から海馬傍回の萎縮が目立つ27).DGでは,AGの出現をはるかに凌駕する領域にpretangleと呼ばれる線維形成性の乏しいタウ陽性細胞の出現を伴っている点が特徴である(図5).嗜銀顆粒は単独に出現して高齢者の認知症の背景病理となるとともに,神経変性疾患に合併して出現する7)30).
3.シヌクレイノパチー
αシヌクレイン(αSN)が脳内に凝集蓄積して封入体を形成する疾患をシヌクレイノパチーと総称する.シヌクレイノパチーの代表的疾患は,レビー小体病(Lewy body disease:LBD)と多系統萎縮症(multiple system atrophy:MSA)であり,neurodegeneration with brain iron accumulation(NBIA)に分類されるPLA2G6の遺伝子変異でもみられる26).1997年,常染色体優性遺伝性PDの家系にαSNの遺伝子変異が発見され,αSN抗体がレビー小体(Lewy bodies:LB)を濃染することからその構成蛋白であることが明らかとなった.αSNは,140個のアミノ酸からなり,主としてシナプス前終末に局在し,脳に豊富に存在している.
1)レビー小体病(LBD)
LBDは,PD,認知症を伴うパーキンソン病(PD with dementia:PDD),DLB,レビー小体を伴う自律神経不全(pure autonomic failure with Lewy bodies:PAF with LB)などを包含する疾患単位である.神経細胞や神経突起にαSN陽性のLB,レビー神経突起(Lewy neurite:LN)を形成することを病理学的特徴とする(図6).
臨床的にパーキンソニズムが先行し,脳幹部の黒質や青斑,迷走神経背側核などの神経細胞脱落,自律神経障害や精神症状を伴う病態がPDである.一方,比較的初期から幻覚,幻視,妄想や変動する意識障害などを呈し,帯状回,扁桃核,海馬傍回などの大脳辺縁系を主体に皮質の小型神経細胞に皮質型LBが出現する病態はDLBと呼ばれ,本邦の小阪らの報告が現在のDLB疾患概念形成の端緒となっている12)13).PDは運動障害を主体とし,DLBは認知症や精神症状が前景になるが,両者はαSN陽性のLB,LNを共通項とする大きなスペクトラムの両極に位置している.2005年のDLBのコンセンサスガイドライン第3版では,病理学的にLBが脳幹部に限局しているタイプ,辺縁系にまで広がっているタイプ,新皮質にびまん性に広がっているタイプの3型に分類されている16).DLBでは病理学的にアルツハイマー病理像を随伴することが多いため,臨床像がDLBの病理像によるものか,ADの病理像によるものかを評価する方式が採用されている.2017年のガイドライン第4版においても第3版の病理学的評価が踏襲されているが,扁桃核や嗅球のLB,LNの有無,黒質の神経細胞脱落の評価が追加されている17).発症1年以内の認知症の出現をDLBとすることが推奨されているが,実際にはパーキンソニズム出現から5年以内の認知症の合併は,DLBの臨床病理像を示す症例が多い.一方,長期経過したPDに認知機能障害が随伴(PDD)すると,病理学的には大脳辺縁系を主体に皮質型LBが広がり,DLBの病理像に達し,PD/DLBの中間的存在として捉えられている.パーキンソニズムが目立たないDLBにおいても黒質や青斑,迷走神経背側核などのαSN陽性LB,LNは観察される.また,PD/DLBでは,全身臓器の末梢自律神経系にもLB,LNが出現することから,全身疾患として理解することが重要である.LBDには,長期にわたり自律神経障害が目立ちパーキンソニズムが顕在化しないPAF with LBの病態が存在し,末梢自律神経系の変性が強い.LBDは頻度の高い疾患であるが,パーキンソニズムや自律神経系の評価は精神科と神経内科では異なっている可能性がある.
2)多系統萎縮症(MSA)
MSAは,病理学的にオリーブ橋小脳萎縮症(olivopontocerebellar atrophy:OPCA)と線条体黒質変性症(striatonigral degeneration:SND),シャイ・ドレーガー症候群(Shy-Drager syndrome:SDS)の3亜型の臨床病理像をとる疾患で,大部分が孤発性疾患であり,オリゴデンドログリアの胞体内にシヌクレイン陽性を示すglial cytoplasmic inclusions(GCI)を形成する22)24)32)(図7).
MSAの国際診断基準では,MSAは排尿障害,インポテンツ,起立性低血圧,発汗低下などの自律神経障害を基盤とし,運動失調を主徴とするMSA-C(MSA with predominant cerebellar ataxia),パーキンソニズムを前景とするMSA-P(MSA with predominant Parkinsonism)の2群に分類し,SDSの呼称は混乱を招くとの理由で推奨されていない8).MSAとLBDは同じシヌクレイノパチーとして分類されるが,LBDは神経細胞の封入体を形成するのに対して,MSAはオリゴデンドログリアにGCIを形成する点で相違があるが,この違いをきたす原因は現時点では未解明である.
4.TDP-43蛋白症
PiDの疾患概念の変遷過程において,臨床的にPiDと区別できないが,ピック球をもたず,タウやシヌクレインに陰性で,ユビキチンにのみ陽性を示す封入体を示す前頭側頭葉変性症(frontotemporal lobar degeneration with ubiquitinated inclusions:FTLD-U)が抽出された28).FTLD-UはしばしばALSを合併することも湯浅,三山らによって報告19)34)されていたが,ユビキチン化された封入体の構成蛋白は長いこと不明であった.2006年にArai, T.,Neumann, M. らによってユビキチン陽性封入体の主要構成蛋白がtrans-activation response(TAR)DNA-binding protein of 43 kDa(TDP-43)蛋白から構成されていることが同定され大きな突破口となった1)23).TDP-43蛋白の凝集,蓄積を基盤とする疾患はTDP-43 proteinopathyと総称される.TDP-43の遺伝子(TAR DNA-binding protein:TARDBP)は,染色体1p36.22上に存在し,414個のアミノ酸から構成され,神経系を含め全身臓器の核内に発現して,遺伝子の転写や選択的スプライシングの調整,mRNAの安定化にかかわるとされている.
1)筋萎縮性側索硬化症(ALS)と前頭側頭葉変性症(FTLD)
ALSの下位運動ニューロンに出現するユビキチン陽性封入体であるskein-like inclusionやround inclusionは,TDP-43免疫染色で陽性を示す(図8).ALSは上位運動ニューロンと下位運動ニューロンの変性を中核とし,運動ニューロン系以外には,通常の病理学的検索では変性が乏しいが,TDP-43免疫染色では,運動ニューロン系を越えて神経細胞内やグリア細胞内に封入体が出現することが明らかになった19)34).
FTLDはタウ蛋白の封入体を伴うFTLD-tauとTDP-43の封入体を伴うFTLD-TDPの2群が大半を占め,いわゆる「ピック球のないPiD」の多くはFTLD-TDPである.FTLD-TDPの病理は封入体の形態と分布をもとにタイプAからタイプDに亜型分類される14)15).タイプAでは皮質II層に多数の神経細胞内封入体(neuronal cytoplasmic inclusions:NCI)と短い変性神経突起(dystrophic neurite:DN)を認め,behavioral variant of FTD(bvFTD)と進行性非流暢性失語症(progressive non-fluent aphasia:PNFA)の臨床像をとる.タイプBでは皮質全層にNCIがみられDNは少数で,bvFTDと運動ニューロン疾患(motor neuron disease:MND)with FTDの臨床像をとる.タイプCでは皮質II層に多数の長いDNを認めNCIは少数であり,語義失語(semantic dementia:SD)とbvFTDの臨床像を示す.タイプDは皮質全層に多数の短いDNと神経細胞核内封入体(neuronal intranuclear inclusion:NII)が出現しNCIは稀である.
欧米のFTLD-TDPの亜型ではタイプAが41%,タイプBが34%,タイプCが25%と報告されている10).本邦のFTLD-TDPではALSを伴うFTLDが多く,タイプBが最も多い.FTLD-TDPの臨床病理像は,前頭側頭型認知症(frontotemporal dementia:FTD)の臨床像が先行してかなり末期にALSの病像が出現する場合もある.認知症が先行する場合には,後期の運動ニューロン障害に気づかれにくい場合もあるため留意が必要である.
表

図1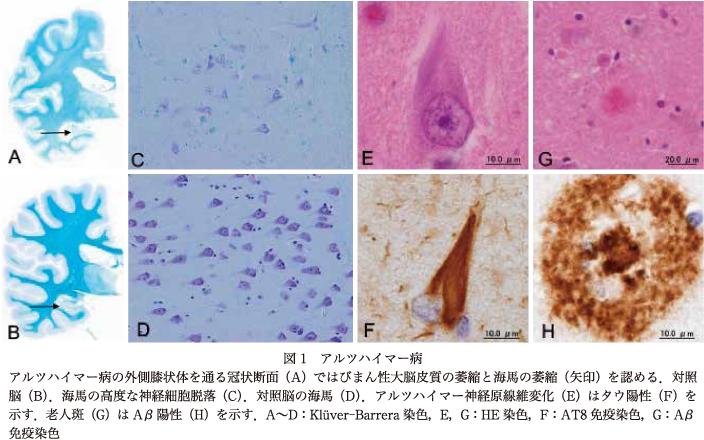

図2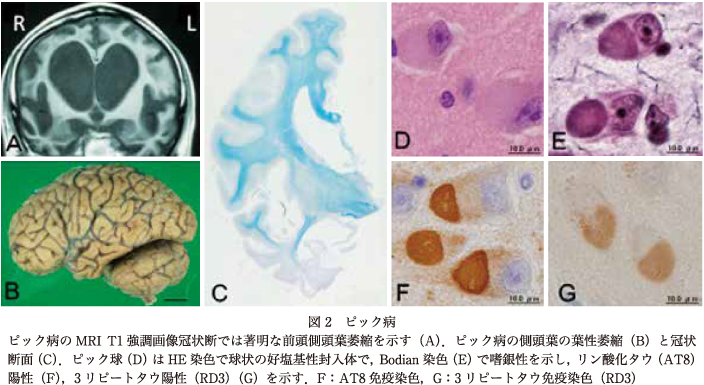

図3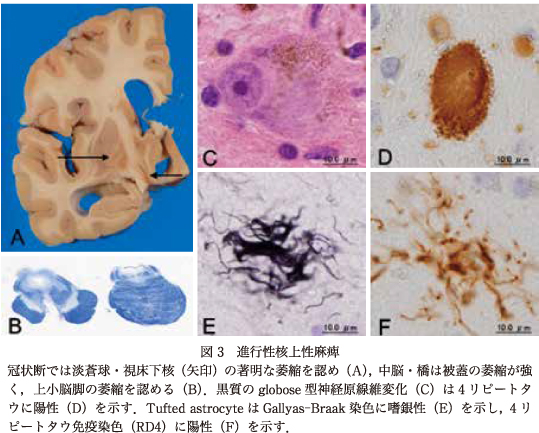

図4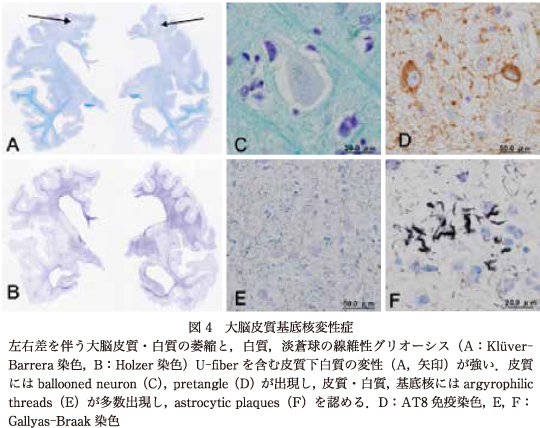

図5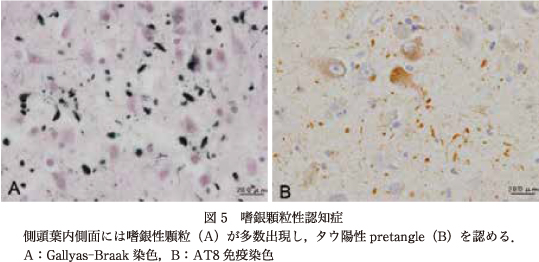

図6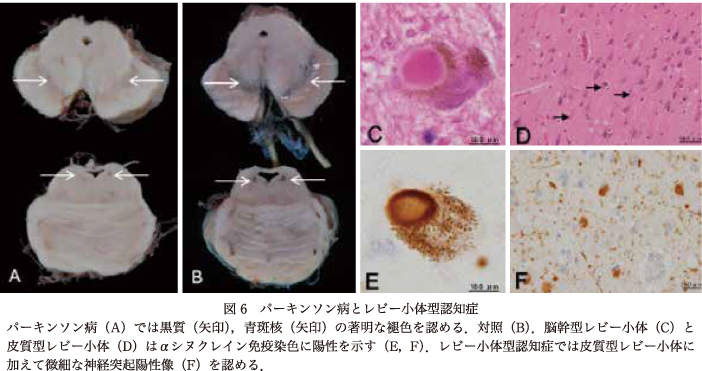

図7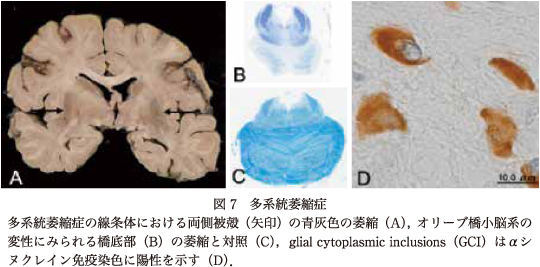

図8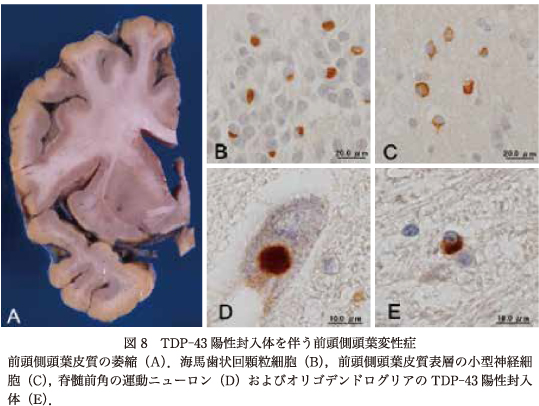

おわりに
認知症などの神経疾患が画像所見などを補助診断として臨床診断される一方,剖検率の低下により神経病理学的検索が激減していることは,極めて憂慮すべき事態である.われわれの施設は年間150例近い剖検脳を検索しているが,臨床診断と病理診断の解離,画像診断では同定できない病理変化などに遭遇し,神経病理学的検索による確定診断の重要性が現代においても意味を失っていないことを日々痛感している.精神科臨床で遭遇する気分障害や,幻覚妄想などは,高齢者においては器質的疾患を背景としていることが少なくない.神経病理学の源流はまさに精神科の先達者に遡ることに改めて敬意を表するとともに,精神科における病理学的検索は今日においても重要であることを強調したい.
本研究の一部は,国立研究開発法人日本医療研究開発機構(AMED)の「脳科学研究推進プログラム」の支援によって行われた.
なお,本論文に関連して開示すべき利益相反はない.
文献
1) Arai, T., Hasegawa, M., Akiyama, H., et al.: TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun, 351; 602-611, 2006![]()
2) Armstrong, M. J., Litvan, I., Lang, A. E., et al.: Criteria for the diagnosis of corticobasal degeneration. Neurology, 80; 496-503, 2013![]()
3) Braak, H., Braak, E.: Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett, 76; 124-127, 1987![]()
4) Braak, H., Braak, E.: Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol, 15; 13-26, 1989![]()
5) Braak, H., Braak, E.: Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol, 82; 239-259, 1991![]()
6) Braak, H., Alafuzoff, I., Arzberger, T., et al.: Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol, 112; 389-404, 2006![]()
7) Ding, Z. T., Wang, Y., Jiang, Y. P., et al.: Argyrophilic grain disease: frequency and neuropathology in centenarians. Acta Neuropathol, 111; 320-328, 2006![]()
8) Gilman, S., Wenning, G. K., Low, P. A., et al.: Second consensus statement on the diagnosis of multiple system atrophy. Neurology, 71; 670-676, 2008![]()
9) Höglinger, G. U., Respondek, G., Stamelou, M., et al.: Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society Criteria. Mov Disord, 32; 853-864, 2017![]()
10) Josephs, K. A., Hodges, J. R., Snowden, J. S., et al.: Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol, 122; 137-153, 2011![]()
11) Komori, T., Arai, N., Oda, M., et al.: Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol, 96; 401-408, 1998![]()
12) Kosaka, K., Oyanagi, S., Matsushita, M., et al.: Presenile dementia with Alzheimer-, Pick- and Lewy-body changes. Acta Neuropathol, 36; 221-233, 1976![]()
13) Kosaka, K.: Diffuse Lewy body disease in Japan. J Neurol, 237; 197-204, 1990![]()
14) Mackenzie, I. R., Neumann, M., Bigio, E. H., et al.: Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol, 119; 1-4, 2010![]()
15) Mackenzie, I. R., Neumann, M., Baborie, A., et al.: A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol, 122; 111-113, 2011![]()
16) McKeith, I. G., Dickson, D. W., Lowe, J., et al.: Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology, 65; 1863-1872, 2005![]()
17) McKeith, I. G., Boeve, B. F., Dickson, D. W., et al.: Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology, 89; 88-100, 2017![]()
18) Mirra, S. S., Heyman, A., McKeel, D., et al.: The consortium to establish a registry for Alzheimer's disease (CERAD). PartII. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology, 41; 479-486, 1991![]()
19) Mitsuyama, Y., Takamiya, S.: Presenile dementia with motor neuron disease in Japan. A new entity? Arch Neurol, 36; 592-593, 1979![]()
20) Montine, T. J., Phelps, C. H., Beach, T. G., et al.: National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease:a practical approach. Acta Neuropathol, 123; 1-11, 2012![]()
21) Murray, M. E., Graff-Radford, N. R., Ross, O. A., et al.: Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol, 10; 785-796, 2011![]()
22) Nakazato, Y., Yamazaki, H., Hirato, J., et al.: Oligodendroglial microtubular tangles in olivopontocerebellar atrophy. J Neuropathol Exp Neurol, 49; 521-530, 1990![]()
23) Neumann, M., Sampathu, D. M., Kwong, L. K., et al.: Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314; 130-133, 2006![]()
24) Papp, M. I., Kahn, J. E., Lantos, P. L.: Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci, 94; 79-100, 1989![]()
25) Rebeiz, J. J., Kolodny, E. H., Richardson, E. P. Jr.: Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc, 92; 23-26, 1967![]()
26) Riku, Y., Ikeuchi, T., Yoshino, H., et al.: Extensive aggregation of α-synuclein and tau in juvenile-onset neuroaxonal dystrophy: an autopsied individual with a novel mutation in the PLA2G6 gene-splicing site. Acta Neuropathol Commun, 1; 12, 2013![]()
27) Saito, Y., Ruberu, N. N., Sawabe, M., et al.: Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol, 63; 911-918, 2004![]()
28) Snowden, J. S., Neary, D., Mann, D. M.: Fronto-temporal Lobar Degeneration: Fronto-temporal Dementia, Progressive Aphasia, Semantic Dementia. Churchill Livingstone, New York, 1996
29) Steele, J. C., Richardson, J. C., Olszewski, J.: Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol, 10; 333-359, 1964![]()
30) Tatsumi, S., Mimuro, M., Iwasaki, Y., et al.: Argyrophilic grains are reliable disease-specific features of corticobasal degeneration. J Neuropathol Exp Neurol, 73; 30-38, 2014![]()
31) Thal, D. R., Rüb, U., Orantes, M., et al.: Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology, 58; 1791-1800, 2002![]()
32) Yoshida, M.: Multiple system atrophy: alpha-synuclein and neuronal degeneration. Neuropathology, 27; 484-493, 2007![]()
33) Yoshida, M.: Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology, 34; 555-570, 2014![]()


