

多くの遺伝子の過剰発現,欠損を伴うコピー数バリエーションは,今までに類をみないほど高率にさまざまな精神疾患と連鎖する.われわれのグループはマウスモデルを使い,22q11.2染色体コピー数欠損,重複と精神疾患の相関に関与するメカニズムの探索に従事してきた.マウスモデルはヒトの染色体上にみられるコピー数欠損,重複の忠実な再現が可能であり,さらにヒトでは不可能な実験的操作によって孤立した変数での因果関係まで探ることを可能にする.遺伝子改変マウスは,脳,神経細胞,神経回路などの機能や構造におけるヒト・マウス間種差が弱点であるが,最終的にヒトに適応される治療の基礎になる推定メカニズムに迫る最短手段としての妥当性は十分に担保されている.われわれのこれまでのデータが示唆する暫定的な推定メカニズムは,22q11.2に含まれている遺伝子のすべてが精神疾患の病態機序構成の要素に関与しているわけではなく,そのなかの特定遺伝子が特定表現型要素に寄与し,遺伝子背景や環境要因によってその発現の有無強度が修飾されるというものである.このメカニズムが他のコピー数バリエーションにも敷衍しうるものかはこれからの研究を俟つ.
第114回日本精神神経学会学術総会
染色体コピー数バリエーションマウスモデルに基づいた精神疾患メカニズムの解体的探索
Albert Einstein College of Medicine, Department of Psychiatry and Behavioral Sciences/Department of Neuroscience/Department of Genetics
精神神経学雑誌
121:
213-223, 2019
<索引用語:精神疾患, マウスモデル, 22q11.2, コピー数バリエーション>
はじめに
われわれの20年近いマウスモデル研究の取り組みを例に,マウスモデルが精神疾患のメカニズムの解明にどれだけ迫りうるかの可能性および,解釈上の問題点について主要な点を概観する.
I.コピー数バリエーションと精神疾患
2008年以来,精神疾患と連鎖した新しいタイプの遺伝子変異が報告されてきている.コピー数バリエーション(copy number variation)と総称されるこれらの遺伝子バリエーションは,相同染色体の片方の領域が数百万塩基にもわたり欠損,あるいは重複したものである.コピー数バリエーションには特筆すべき3つの特性がある.第一に,精神疾患の集団からみると,コピー数バリエーションは稀であり,どの精神疾患の集団でも1%以下で存在する.多くは0.3%あるいはそれ以下の頻度である.例えば,統合失調症集団では,22q11.2欠損は0.3%以下の頻度で存在する29)38).1995年の報告で100名の統合失調症患者のうち2名が22q11.2欠損をもつことから算出された2%の数値がそのグループによっていまだに主張され,その主張を孫引きしているものが散見される.また,別のグループも459名の統合失調症と統合失調症感情障害の患者のなかで,5名の22q11欠損が見つかったことから,1%という数値を主張している.しかし,最近の12,202名あるいは19,084名の統合失調症ケースに基づく大規模研究で,22q11.2欠損は0.3%あるいはそれ以下と報告され,1%や2%という頻度は否定されている29)30)38).
第二に,各コピー数バリエーション集団からみると,今までに類がないレベルの精神疾患発現率が認められる.各コピー数バリエーションをもつことで,どれだけ疾病罹患率があるかを示す浸透率(penetrance)は,統合失調症では3q29欠損で15%以上,22q11欠損で10%以上,7q11.23重複,16q11.2重複で5%以上と一般人口での罹患率をはるかに上回る.自閉スペクトラム症,発達遅延および何らかの身体的発達遅延を含めた発達障害集団での浸透率は7q11.23欠損でほぼ100%,22q11欠損で90%,15q11-13重複,1q21.1欠損,15q13.3欠損,16p11.2欠損でも30%以上となっている.
第三に,各コピー数バリエーションは統合失調症,自閉スペクトラム症,知的能力障害,双極性障害,再発性うつ病,ADHDなど診断上別個とみなされる多岐にわたる精神疾患に連鎖している13)29)30).このことは,臨床上別個の診断とみなされているものが同じ遺伝的バリエーションから引き起こされることを意味し,臨床基準に基づく分類は,分子生物学的メカニズムの観点からは支持されない可能性を提示している.
この第三の点に関して,各疾病の症状の全体像を単位として生物学的病態機序のメカニズムを求めることに無理があることは,これまでにもいくつかの論拠から指摘されてきた.同じ精神疾患でも各症状の出現には個体間で差があり,疾病のすべての構成要素の症状が同程度に出ているわけではない.また,臨床上の診断基準を満たして疾病と確定診断される以前にも,いろいろな非定型要素が出現する.統合失調症の診断がつく以前より認知機能の低下が先行することはよく知られている.自閉スペクトラム症では,社会場面における注意欠如,非定型な泣き声,顔のなかの特に目に対する注意欠如など,診断基準に入っていない,あるいは入っていても診断に至るほど重篤ではない要素も,一般の診断基準で疾病と診断される前から顕在化している.遺伝的バリエーションが,臨床診断時に突然起こることはあり得ず,脳内機能は診断以前から非定型的に何らかの障害を引き起こしていると想定される.現在の精神疾患分類上の疾病は便宜的に決められた閾値を超えたために,臨床上,ある特定の疾病として類型診断されたものであり,それに基づく各疾病研究は,漸次的に出現する疾病に連鎖した脳内メカニズム探索と相容れないものであることは明らかである.
さらに,疾病診断上の構成要素が,一律に治療薬に反応しないことから,各症状のメカニズムの多様性が示唆されてきた.例えば,定型抗精神病薬ハロペリドールは,統合失調症の妄想や幻覚には効果があるが,やる気のなさ,社会的な回避,認知機能低下には効果がない.また,非定型抗精神病薬のリスペリドン,オランザピン,アリピプラゾールなどは,自閉スペクトラム症の攻撃性,常同行動をある程度抑えるが,社会行動の非定型さには効果がない.
1つ1つのコピー数バリエーションが多岐にわたる精神疾患と連鎖していることは,遺伝子,分子経路メカニズムの観点から,多くの精神疾患が共通の基盤をもったものであることを示唆し,従来の精神疾患単位でメカニズムを追う方法論の妥当性に疑問を投げかけている.同時に,明らかに各疾患は見かけ上異なる症状・構成要素のまとまりとして臨床上,認識されており,同じ生物学的基盤から多岐にわたる表現型がどのようにして現れるかはいまだわかっていない.
II.22q11.2コピー数バリエーション
われわれのグループはヒト染色体22q11コピー数バリエーションの解明に従事してきた25).この染色体バリエーションと精神疾患との連鎖は興味深い歴史的変遷をたどっている.
Philadelphia, Temple大学の小児内分泌専門医Angelo DiGeorgeは,胸腺の欠如あるいは形成不全と副甲状腺縮小による副甲状腺ホルモン低下,円錐動脈幹異常などの先天性心疾患を主症状とする,後にDiGeorge syndromeと呼ばれる患者群を1960年代から報告していた.
New York,Albert Einstein College of Medicineとその附属病院Montefiore Hospitalの言語病理専門家R. J. Shprintzenは粘膜下口蓋裂や口蓋裂などの鼻咽腔閉鎖機能不全,先天性心疾患,小さく低い位置にある耳,離れた目の位置,厚めのまぶた,比較的長い顔,短い上唇などで特徴づけられる典型的な顔の骨格,筋緊張低下,学習困難を呈する一群の患者を1978年に報告した41).この症候群は後にvelo-cardio-facial syndrome(VCFS)と呼ばれた42).
Shprintzenらは,この症候群の患者が成人するにつれて10%を超える率で統合失調症の診断を下されていることを1992年に報告し43),同年VCFSの患者は一様に染色体22q11欠損があることを報告したことで10)39),遺伝子で同定可能な統合失調症の一群が初めて発見されたのである.同年,DiGeorge syndromeも22q11欠損をもつことが報告され7)9),VCFSとDiGeorge syndromeの見かけ上の差は,単に症候群内の各症状の個人間でのばらつきと担当医がどの症状に注目したかを反映するものであることが明らかとなった.
最近の大規模サンプルを使った研究では,成人22q11.2欠損保持者のうち30%が統合失調症と診断されることが明らかになっている40).年齢群によってばらつきがあるものの,22q11.2欠損では自閉スペクトラム症(13~27%),注意欠如・多動症(16~36%),うつ病(2~16%),不安症(24~36%)も高率に診断される1)15)27)32)40)52).
欠損だけでなく,Albert Einstein College of Medicineの研究チームによって発見された22q11重複患者では12),てんかん,知的能力障害,注意欠如・多動症,自閉スペクトラム症がみられるが35),現段階ではサンプル数が少なく正確な頻度を示すことはできない.22q11重複は統合失調症になりにくい要因と考えられてきたが37),最近,偏執妄想,混沌とした思考,幻覚,自殺念慮,感情症状などを呈するケースが報告されている50)53).また,22q11重複保持者104名中,統合失調症患者が4名みられるという報告もあることから35),今後より大きなサンプルでの解析を俟つ.
III.マウスモデルの必要性
精神疾患に寄与する脳内機序の解明は現在の技術ではヒトでは困難である.血液内の細胞で起こっている遺伝子の発現,修飾が脳内での変化とどれだけ相関しているかは不明である.ヒトでは,コピー数バリエーションと精神疾患の相関は明確であるが,コピー数バリエーションと脳画像や死後脳での変化の相関は因果関係を特定するものではない.
そこで因果関係の同定を可能にするモデルが必要となる.ヒトinduced pluripotent stem cell(iPS細胞)は唯一,精神疾患をもつヒトの神経細胞の培養を可能にする手段であるが,作製された神経細胞は,ヒトにおける受精後16週齢までの神経細胞と比較して遺伝子発現パターンで部分的類似性が認められるものの,それ以降の週齢ではあまり類似性が認められない6).iPS細胞から作製された胎児様神経細胞の異常が,精神疾患発現に因果関係をもつメカニズムの解明あるいは治療薬開発にどれだけ意味があるモデルを提供しうるか現時点では不明である.回路に組み込まれていないiPS細胞由来の神経細胞で精神疾患に特有な異常を解明することができるかもまた不明である.さらに,iPS細胞由来神経細胞は表現型のばらつきが大きく,現段階では統制群と疾病群の比較に再現性が高くないケースが多い.脳organoidも初期発達のモデル化には適しているが,脳の発達段階での変化,また環境要因との相互作用を解明することはできない.また,現時点でのプロトコールでは同じ培養条件でもorganoid間の均質性が保たれていない.これらの手法はモデルとしての再現性の点でまだ技術面で改善の余地があり,さらにモデル自体の妥当性の面からも解釈上注意すべき点が多い.
22q11の個々の遺伝子欠損モデルは,マウスのほかに,Zebrafish,Drosophila,C. Elegansがあり,心臓,頭蓋顔面,脳の形成などに関与した遺伝子の同定に一定の成功を収めている17).しかし,これらのモデルは精神疾患の脳内メカニズム解明には多くの問題点を抱えている.遺伝子の面からは,22q11に含まれている遺伝子群が1箇所の染色体にまとまっておらずセグメント単位の欠損,重複モデル作製が困難であったり,ヒト22q11遺伝子に匹敵する遺伝子がこれらのモデルでは見つかっていない場合もある.ヒト22q11.2の蛋白生成遺伝子のうち,同定されているものはDrosophilaでは48%,C. Elegansでは37%にとどまる.ヒト22q11でのマイクロRNAにいたってはモデル動物ではマウス,Zebrafishでの数件を除いて見つかっていない.さらに,脳神経系の構造に哺乳類と比較して顕著な種間差があり,脳内の解析対象部位の哺乳類との関連が不明瞭である.行動面からは,高度認知機能など哺乳類に特有の行動がモデル化できない場合があり,しかも発達上の行動成熟が哺乳類と違うことから発達上での変化を尺度として解析対象とすることも困難である.したがって,これらのモデルは精神疾患に関与しないにもかかわらず行動表現型が検出される偽陽性(false positive),あるいは精神疾患への関与があるにもかかわらず関連行動表現型が検出できない偽陰性(false negative)の可能性がマウスよりも大きく,モデルとしての妥当性が現段階では不明瞭である.しかし,より複雑な社会行動などの行動解析がメダカで行われており51)55),遺伝子操作および大規模スクリーニングの容易さから,メダカやZebrafishにはモデルとしての妥当性がありさらなる発展の余地がある.
多くの場合,ヒトコピー数バリエーションに内包されている遺伝子はマウスゲノムにもあることから,コピー数バリエーションはマウスモデルで精神疾患脳内メカニズムを解明するための盤石な出発点である.しかし,ヒトコピー数バリエーションを,なぜマウスに人為的に作製する必要があるのであろうか.ヒトコピー数バリエーションはほとんどの場合多くの遺伝子を含んでおり,それらがどのように精神疾患に関与するかをヒトでは同定できない.1つの極端な仮説として,ヒトコピー数バリエーションに含まれているすべての遺伝子が総和として精神疾患に寄与する可能性がある.この仮説はさらに,すべての遺伝子が同等に各精神疾患に関与する可能性と,すべての遺伝子が精神疾患の異なる要素に関与する可能性に細分される.別の極端な考え方として,単一遺伝子あるいは一部の遺伝子のみが関与すると仮定することも可能である.一部の原因遺伝子の寄与度は疾患要素の種類によって異なる可能性もある.ヒトでは,これらの仮説の検証実験は不可能である.一方,マウスモデルでは,コピー数バリエーションの一部あるいは単一遺伝子の欠損,重複モデルから,どの部位あるいは遺伝子が,疾患のどの要素に寄与するかを実験的に検証することが可能である.さらに,脳内での神経細胞の変化を詳細に検証することもマウスモデルでは可能である.
もちろん,マウスを含むモデル動物で精神疾患を忠実に再現することはできない.幻聴,妄想,話の支離滅裂さなどの統合失調症の診断要素はモデル動物において存在せず,たとえ類似の表現型があったとしても客観的にそれらを測定することは不可能である.また,モデル動物の社会行動,認知機能がヒトと同一の脳部位,分子に依存しているという前提も明確な根拠があるわけではない.例えば,自閉スペクトラム症構成要素である社会行動は,イヌやマウスでは嗅覚に依存するが,ヒトでは視聴覚に依存する.このことから,モデル動物での知見は暫定的,仮説(推定)的メカニズムであるという認識が解釈上重要である.
IV.精神疾患と基軸次元
最近提唱された基軸次元(dimension)31)に注目する精神疾患の解析は,複数の精神疾患で非定型である各疾病に特異性のない行動,指標を解析対象とする.例えば,作業記憶の低下は,統合失調症,注意欠如・多動症,自閉スペクトラム症などでみられ,1つの精神疾患に特定されるものではない.ヒトでは,理由は微妙に異なるが,社会行動の低下は自閉スペクトラム症だけでなく,統合失調症,うつ病,注意欠如・多動症などでみられる.マウスの社会行動からはそういった違いを弁別することは不可能であり,純粋に疾病とは独立した基軸次元の1つとして扱う以外に手がない.疾病単位とは独立に変異する他の行動基軸次元に,知覚やその処理,プレパルス抑制などがある.
遺伝子変異マウスモデルを使った脳機能あるいは細胞分子レベルでの解析が,精神疾患のよりよい理解に直結することは容易に抱ける楽観的展望であろうが,落とし穴もみえてきた.行動表現型を無視して,遺伝子変異によって惹起される脳内変化だけを探しても,それがすべてメカニズムになる可能性は支持されていない.個々の行動表現型に影響を及ぼす遺伝子は各染色体コピー数バリエーションの一部であることが実験的に示されている23)26).また,別々の行動表現型に関与している染色体コピー数バリエーション内の遺伝子が同一のセットではないことから,多くの遺伝子が何らかの症状の側面に寄与したとしても,その作用機序は複数の細胞レベルでのメカニズムが想定されている.例えば染色体コピー数バリエーションに含まれているある遺伝子の欠損によって,シナプスあるいは細胞に電気生理学的変化が起こったとしても,行動表現型としては何の変化もなかった場合,2つの可能性がある.1つは,それらの細胞表現型は今まで測定されていない行動表現型のメカニズムである可能性.この場合,より広範な行動解析で相関する行動表現型が見つかる可能性が残される.それでも,行動表現型でまったく異常が見つからない場合は,出現した細胞,シナプス表現型は,いまだ行動表現型が見つかっていないものに関与しているか,あるいは行動異常を起こさない付帯表現型(epi-phenotype)である可能性がある26).どの遺伝子の欠損,重複でも脳内で何らかの遺伝子発現,細胞表現型を引き起こすが,それらのすべてが症状の原因ではないことから,何らかの行動表現型との相関をみることなく,遺伝子変異と脳内表現型だけに着目した研究は,疑似メカニズムを標的にした治療の開発の基になりかねない.
遺伝子変異がいくつかの基軸次元(行動,細胞,発達)に基づいた複合的立体的空間をどのように歪めるかをマウスモデルによって再構築していくことが,精神疾患をメカニズムの観点から解明し,メカニズムに基づいた治療開発へつなげる1つの有効な戦略であることが示唆できる.
V.22q11.2の欠損モデルから推察される行動表現型の脳内メカニズム
多くのコピー数バリエーションがそれまでに類をみないほど高率でさまざまな精神疾患と連鎖していることが2008年からの一連の研究によって明らかとなり,そこからマウスモデルの作製,解析が始まった.22q11コピー数バリエーションはその発見の経緯から他のコピー数バリエーションよりはるかに先行したため,多くのマウスモデルが作製,解析されてきている22-24)26).
1999年にW. L. Kimberらは,Zip520-psからSlc25a1までの区域を欠損させたマウスが,22q11欠損患者とは真逆で,プレパルス抑制亢進を示すことを報告した28).このことから,22q11は全体の欠損とは真逆の効果を及ぼす部分を内包していることが示唆される23).
われわれのチームは,別の領域200 kbを過剰発現させたマウスは強迫的常同的運動を示し,正常な社会行動が不可能だが,3週間クロザピンを投与すると常同的行動異常が軽減することを報告した21).この部位はGnb1l,Tbx1,Gp1bb,Sept5を含むことから,これらのうち単一,あるいは複数の遺伝子がこの行動異常に関与している可能性があった.
R. Paylorらは,われわれの同定した200 kbの欠損が含まれたときのみ大きな染色体欠損でプレパルス抑制が低下することをマウスモデルで示した36).Paylorらの研究でさらに重要な点は,この200 kbの外で欠損を起こしても,プレパルス抑制は低下しなかったことである.プレパルス抑制に何ら変化を及ぼさなかった染色体領域には,Prodh,Zdhhc8,Ranbp1,Dgcr8,Mrpl40,Hira,Comtなどが含まれる.後続研究で,これらの遺伝子がさまざまな脳内表現型に関与していることが報告されているが,それら脳内表現型は少なくともプレパルス抑制に限っていえば,因果関係のある脳内メカニズムと結論することには問題がある22)23)26).
われわれはさらに,この200 kb領域外でも,別の基軸次元に関与している部位を同定した.ARVCF,COMT,TXNRD2を含む190 kb隣接領域を過剰発現したマウスでは,プレパルス抑制や社会行動では異常は起きないが,作業記憶に限局した異常が起こる45).さらに,この表現型は作業記憶自体が異常になったのではなく,発達上幼児期から成人期にかけての作業記憶拡大の度合いが思春期で頭打ちになる結果であることもわれわれの研究は示した45).その後発表された研究で,COMTの高機能一塩基多型をもつ健常者でも同様に,発達上の作業記憶拡大が思春期以降頭打ちになることが報告された11).このことから,われわれが同定した190 kb領域に含まれる3遺伝子のうち少なくともCOMTはこの表現型に関与している可能性がある.
われわれは,さらにこれら190 kbと200 kb領域中で行動表現型の原因遺伝子を同定する作業に研究対象を絞り込んでいった.Sept5は,シナプスからのドパミンやグルタミン酸神経伝達分子の放出を抑制制御することや3)4)8)54),神経軸索および複雑な樹状突起の形成に関与している46)49).さらに,SEPT5とGB1BBの欠損患者が1例報告され,この患者は発達遅延が認められた2).われわれの解析では,Sept5ヘテロマウスでは行動上の異常はみられなかったが,ホモマウスにおいて相互社会行動と報酬に対して接近行動を学習するincentive learningの低下,プレパルス抑制亢進および不安行動低下がみられた44).プレパルス抑制および不安に関連した行動異常は22q11.2欠損患者とは真逆の表現型であることから,22q11欠損の最終表現型は含まれている多くの遺伝子の真逆なあるいは同一方向の表現型の総和としてあらわれることが示唆される23).また,Sept5ホモマウスでの社会行動低下は22q11欠損患者と同一方向の表現型であるが,実験的に遺伝子背景を変えることで表現型の度合いが大きく変わることが示された18)22)44).このことは,ゲノム全体での一塩基多型の集合体としての背景遺伝子が22q11に含まれている遺伝子欠損の表現型を大きく修飾することを意味し,ヒトにおいてみられる22q11欠損に付随する精神疾患診断の多様性の説明概念とみなすことが可能である22)23).また,Sept5はその海馬での遺伝子量によって相互社会行動のレベルが決定された.扁桃体でのSept5発現量は環境要因によって変化することから18),Sept5は環境要因が行動表現型を修飾するときに介在する一遺伝子であることが示唆される.
200 kbに含まれているもう1つの遺伝子である転写因子TBX1の変異はヒトで数例報告されている.TBX1欠損を起こすexon9でのinsertionあるいはframeshift deletionは自閉スペクトラム症を発症するケースがある16)34)36).TBX1の遺伝子発現を阻害するexon8のframeshift duplicationをもつ家系ではてんかん患者がみられる19).TBX1を過剰発現するmissense mutationや一塩基多型も知的能力障害の症状を伴う48)56).
これらのヒトでの連鎖は単なる相関であり,他の一塩基多型やmutationが真の原因であることも否定できない.実際,TBX1 mutation保持者は他遺伝子のmutationや一塩基多型をもっている34).そこで,われわれは遺伝子背景を統制したcongenic Tbx1ヘテロマウスを作製し,広範な行動解析を行った.この結果,Tbx1ヘテロマウスは生まれてから8日ですでに母親との社会コミュニケーション機能が低下しており20)47),思春期以降の行動でも社会行動の低下,社会的でない物体への興味亢進,作業記憶の低下と柔軟性のなさなどを認めた20).
特に,幼児期社会コミュニケーションの機能低下は顕著で,ヘテロマウスの鳴き声は母マウスの母性行動を誘引せず,その原因は発声される鳴き声の構造変化によることが判明した47).ヘテロマウスは2ヵ月齢(マウス成人年齢)で相互社会行動が著しく低いことが観察されたことから20),マウスにおける2ヵ月齢での行動異常の予測が生後1週間でできることを示唆する.ヒトにおいてものちに自閉スペクトラム症と診断された赤ちゃんの泣き声の非定型さが知られており14),この初期症状がのちの自閉スペクトラム症診断の予測に使えることが示唆されている14).少なくとも,マウスモデルにおいてはこの予測は担保されていることから,ヒトでも早い診断,治療の一助となることが期待される.
過剰発現された190 kb領域の部位においては,COMTのアミノ酸置換を伴う高機能性一塩基多型が,発達過程での作業記憶容量拡大の頭打ちと連鎖していることが知られているが11),その作用脳部位,細胞はわかっていなかった.Tbx1を含む200 kbのマウスでの過剰発現も行動異常を引き起こし21),しかもTbx1蛋白は成体神経幹細胞で高レベルの発現をみせていることから20),われわれはTbx1とCOMTをマウスの成体神経幹細胞に選択的に発現させて行動解析を行った.成体神経幹細胞でこれらの遺伝子を過剰発現させると,どちらでも発達上での作業記憶容量拡大が抑えられることが確認された5).また,この研究で,どちらの遺伝子を成体神経幹細胞で過剰発現させても,幹細胞が顆粒細胞層のなかを分化移動しにくくなっていることも明らかとなった5).このことから,作業記憶収容量の発達上拡大の抑制という表現型に限っていえば,この2つの22q11.2遺伝子は成体神経幹細胞だけでの過剰発現を通して再現できることがわかった.しかし,このことは他の細胞群で同様な寄与が起こらないことは意味せず,さらなる解析を俟つ.
おわりに
マウスモデルを使った研究から,22q11.2コピー数バリエーションに含まれるすべての遺伝子が均等に基軸次元に関与せず,特定遺伝子が特定基軸次元に発達上時間軸で影響を及ぼし,その影響も遺伝子背景や環境要因により修飾されることが示唆されてきている.この総説ではTbx1やCOMTに焦点をあて,22q11に含まれる遺伝子が行動と発達軌跡の基軸次元上どう関連しているかを明らかにした(図).他の単一遺伝子の解析と合わせ,22q11.2に含まれる遺伝子が多くの場合,脳内表現型を引き起こすにもかかわらず行動表現型を引き起こさないことが明らかとなり,コピー数バリエーション全域のモデルマウスでは,精神疾患とは因果関係のない脳内表現型までもメカニズムと誤認する可能性がある.マウスモデルでのコピー数バリエーションモデルの部分部分あるいは単一遺伝子モデルの解析によってこの解釈上の落とし穴を回避することが可能であるが,単純なヒトコピー数バリエーションモデルでの遺伝子発現変異やヒトでのbiological pathway解析では,症状とは関係ない遺伝子群も含んでいる可能性があり,そのようなデータに基づく精神疾患の理解,治療薬の開発は問題がある.マウスデータが示唆する精神疾患の理解,治療薬の開発への妥当なアプローチは,コピー数バリエーションに含まれる遺伝子レベルでの解体的探索と,多次元表現型基軸に基づく遺伝子と行動表現型間の因果関係の再構築である.
図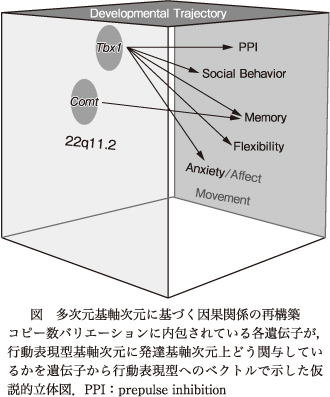

この論文は以下の研究助成金の援助のもと作成された.R01MH099660,R01DC015776,R21HD053114,U54HD090260
第114回日本精神神経学会学術総会=会期:2018年6月21~23日,会場=神戸国際会議場,神戸国際展示場,神戸ポートピアホテル
総会基本テーマ:精神医学・医療の普遍性と独自性―医学・医療の変革のなかで―
教育講演:染色体コピー数バリエーションマウスモデルに基づいた精神疾患メカニズムの解体的探索
座長:朴 秀賢(神戸大学大学院医学研究科精神医学分野)
なお,本論文に関連して開示すべき利益相反はない.
謝 辞 この論文は第114回日本精神神経学会学術総会での教育講演に基づいている.この教育講演を提案企画していただいた大阪医科大学の米田博先生,金沢徹文先生に感謝するとともに,金沢徹文先生および神戸大学精神科の菱本明豊先生には原稿の内容に関して多くのコメントをいただいたことをここに特に謝す.ここで議論された研究に携わった廣井研究室の朴秀賢先生,平本豪先生,高橋知久先生,鈴木豪先生,Kathryn Harper博士,吾妻壮先生,泉剛先生,菱本明豊先生,阿部誠治先生,澤村岳人先生,榎本真悟先生,西晃先生,能丸寛子先生,永嶋雅子先生,中康彦先生に謝辞を表する.論文の校正にあたった廣井元子氏に感謝する.
文献
1) Antshel, K. M., Aneja, A., Strunge, L., et al.: Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion). J Autism Dev Disord, 37 (9); 1776-1786, 2007![]()
2) Bartsch, I., Sandrock, K., Lanza, F., et al.: Deletion of human GP1BB and SEPT5 is associated with Bernard-Soulier syndrome, platelet secretion defect, polymicrogyria, and developmental delay. Thromb Haemost, 106 (3); 475-483, 2011![]()
3) Beites, C. L., Xie, H., Bowser, R., et al.: The septin CDCrel-1 binds syntaxin and inhibits exocytosis. Nat Neurosci, 2 (5); 434-439, 1999![]()
4) Beites, C. L., Peng, X. R., Trimble, W. S.: Expression and analysis of properties of septin CDCrel-1 in exocytosis. Methods Enzymol, 329; 499-510, 2001![]()
5) Boku, S., Izumi, T., Abe, S., et al.: Copy number elevation of 22q11.2 genes arrests the developmental maturation of working memory capacity and adult hippocampal neurogenesis. Mol Psychiatry, 23; 985-992, 2018![]()
6) Brennand, K., Savas, J. N., Kim, Y., et al.: Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry, 20 (3); 361-368, 2015![]()
7) Carey, A. H., Claussen, U., Lüdecke, H. J., et al.: Interstitial deletions in DiGeorge syndrome detected with microclones from 22q11. Mamm Genome, 3; 101-105, 1992![]()
8) Dong, Z., Ferger, B., Paterna, J. C., et al.: Dopamine-dependent neurodegeneration in rats induced by viral vector-mediated overexpression of the parkin target protein, CDCrel-1. Proc Natl Acad Sci U S A, 100 (21); 12438-12443, 2003![]()
9) Driscoll, D. A., Budarf, M. L., Emanuel, B. S.: A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet, 50 (5); 924-933, 1992![]()
10) Driscoll, D. A., Spinner, N. B., Budarf, M. L., et al.: Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am J Med Genet, 44; 261-268, 1992![]()
11) Dumontheil, I., Roggeman, C., Ziermans, T., et al.: Influence of the COMT genotype on working memory and brain activity changes during development. Biol Psychiatry, 70 (3); 222-229, 2011![]()
12) Edelmann, L., Pandita, R. K., Spiteri, E., et al.: A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet, 8 (7); 1157-1167, 1999![]()
13) Elia, J., Glessner, J. T., Wang, K., et al.: Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet, 44 (1); 78-84, 2011![]()
14) Esposito, G., Hiroi, N., Scattoni, M. L.: Cry, baby, cry: expression of distress as a biomarker and modulator in autism spectrum disorder. Int J Neuropsychopharmacol, 20 (6); 498-503, 2017![]()
15) Fine, S. E., Weissman, A., Gerdes, M., et al.: Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J Autism Dev Disord, 35; 461-470, 2005![]()
16) Gong, W., Gottlieb, S., Collins, J., et al.: Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet, 38 (12); E45, 2001![]()
17) Guna, A., Butcher, N. J., Bassett, A. S.: Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J Neurodev Disord, 7 (1); 18, 2015![]()
18) Harper, K. M., Hiramoto, T., Tanigaki, K., et al.: Alterations of social interaction through genetic and environmental manipulation of the 22q11.2 gene Sept5 in the mouse brain. Hum Mol Genet, 21; 3489-3499, 2012![]()
19) Hasegawa, K., Tanaka, H., Higuchi, Y., et al.: Novel heterozygous mutation in TBX1 in an infant with hypocalcemic seizures. Clin Pediatr Endocrinol, 27; 159-164, 2018![]()
20) Hiramoto, T., Kang, G., Suzuki, G., et al.: Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum Mol Genet, 20; 4775-4785, 2011![]()
21) Hiroi, N., Zhu, H., Lee, M., et al.: A 200-kb region of human chromosome 22q11.2 confers antipsychotic-responsive behavioral abnormalities in mice. Proc Natl Acad Sci U S A, 102 (52); 19132-19137, 2005![]()
22) Hiroi, N., Hiramoto, T., Harper, K. M., et al.: Mouse models of 22q11.2-associated autism spectrum disorder. Autism, S1; 1-9, 2012![]()
23) Hiroi, N., Takahashi, T., Hishimoto, A., et al.: Copy number variation at 22q11.2: from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol Psychiatry, 18 (11); 1153-1165, 2013![]()
24) Hiroi, N., Nishi, A.: Dimensional deconstruction and reconstruction of CNV-associated neuropsychiatric disorders. Modeling the Psychopathological Dimensions of Schizophrenia: From Molecules to Behavior (ed by Pletnikov, M., Waddington, J.). Academic Press/Elsevier, London, p.285-302, 2016
25) 廣井 昇, 吉川 武男: 22q11.2欠失症候群および22q11.2重複症候群. 脳科学辞典. 2016 (https://bsd.neuroinf.jp/wiki/22q11.2欠失症候群および22q11.2重複症候群)(参照2018-12-19)
26) Hiroi, N.: Critical reappraisal of mechanistic links of copy number variants to dimensional constructs of neuropsychiatric disorders in mouse models. Psychiatry Clin Neurosci, 72; 301-321, 2018![]()
27) Kates, W. R., Antshel, K. M., Fremont, W. P., et al.: Comparing phenotypes in patients with idiopathic autism to patients with velocardiofacial syndrome (22q11 DS) with and without autism. Am J Med Genet A, 143A (22); 2642-2650, 2007![]()
28) Kimber, W. L., Hsieh, P., Hirotsune, S., et al.: Deletion of 150 kb in the minimal DiGeorge/velocardiofacial syndrome critical region in mouse. Hum Mol Genet, 8 (12); 2229-2237, 1999![]()
29) Kirov, G., Rees, E., Walters, J. T., et al.: The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry, 75 (5); 378-385, 2014![]()
30) Malhotra, D., Sebat, J.: CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell, 148 (6); 1223-1241, 2012![]()
31) Morris, S. E., Cuthbert, B. N.: Research domain criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci, 14 (1); 29-37, 2012![]()
32) Niklasson, L., Rasmussen, P., Oskarsdóttir, S., et al.: Chromosome 22q11 deletion syndrome (CATCH 22): neuropsychiatric and neuropsychological aspects. Dev Med Child Neurol, 44 (1); 44-50, 2002![]()
33) Nishi, A., Hiroi, N.: Genetic mechanisms emerging from mouse models of CNV-associated neuropsychiatric disorders. The Neurobiology of Schizophrenia (ed by Abel, T., Nickl-Jockschat, T.). Academic Press/Elsevier, New York, p.397-417, 2016
34) Ogata, T., Niihori, T., Tanaka, N., et al.: TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia. PLoS One, 9 (3); e91598, 2014![]()
35) Olsen, L., Sparsø, T., Weinsheimer, S. M., et al.: Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry, 5 (7); 573-580, 2018![]()
36) Paylor, R., Glaser, B., Mupo, A., et al.: Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci U S A, 103 (20); 7729-7734, 2006![]()
37) Rees, E., Kirov, G., Sanders, A., et al.: Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry, 19 (1); 37-40, 2014![]()
38) Rees, E., Walters, J. T., Georgieva, L., et al.: Analysis of copy number variations at 15 schizophrenia-associated loci. Br J Psychiatry, 204 (2); 108-114, 2014![]()
39) Scambler, P. J., Kelly, D., Lindsay, E., et al.: Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet, 339 (8802); 1138-1139, 1992![]()
40) Schneider, M., Debbané, M., Bassett, A. S., et al.: Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the international consortium on brain and behavior in 22q11.2 deletion syndrome. Am J Psychiatry, 171 (6); 627-639, 2014![]()
41) Shprintzen, R. J., Goldberg, R. B., Lewin, M. L., et al.: A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J, 15 (1); 56-62, 1978![]()
42) Shprintzen, R. J., Goldberg, R. B., Young, D., et al.: The velo-cardio-facial syndrome: a clinical and genetic analysis. Pediatrics, 67 (2); 167-172, 1981![]()
43) Shprintzen, R. J., Goldberg, R., Golding-Kushner, K. J., et al.: Late-onset psychosis in the velo-cardio-facial syndrome. Am J Med Genet, 42 (1); 141-142, 1992![]()
44) Suzuki, G., Harper, K. M., Hiramoto, T., et al.: Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Hum Mol Genet, 18 (9); 1652-1660, 2009![]()
45) Suzuki, G., Harper, K. M., Hiramoto, T., et al.: Over-expression of a human chromosome 22q11.2 segment including TXNRD2, COMT and ARVCF developmentally affects incentive learning and working memory in mice. Hum Mol Genet, 18 (20); 3914-3925, 2009![]()
46) Tada, T., Simonetta, A., Batterton, M., et al.: Role of Septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr Biol, 17 (20); 1752-1758, 2007![]()
47) Takahashi, T., Okabe, S., Broin, P. Ó., et al.: Structure and function of neonatal social communication in a genetic mouse model of autism. Mol Psychiatry, 21 (9); 1208-1214, 2016![]()
48) Torres-Juan, L., Rosell, J., Morla, M., et al.: Mutations in TBX1 genocopy the 22q11.2 deletion and duplication syndromes: a new susceptibility factor for mental retardation. Eur J Hum Genet, 15 (6); 658-663, 2007![]()
49) Tsang, C. W., Estey, M. P., DiCiccio, J. E., et al.: Characterization of presynaptic septin complexes in mammalian hippocampal neurons. Biol Chem, 392 (8-9); 739-749, 2011![]()
50) van Amelsvoort, T., Denayer, A., Boermans, J., et al.: Psychotic disorder associated with 22q11.2 duplication syndrome. Psychiatry Res, 236; 206-207, 2016![]()
51) Wang, M. Y., Takeuchi, H.: Individual recognition and the 'face inversion effect' in medaka fish (Oryzias latipes). eLIFE, 6; e24728, 2017![]()
52) Wenger, T. L., Miller, J. S., DePolo, L. M., et al.: 22q11.2 duplication syndrome: elevated rate of autism spectrum disorder and need for medical screening. Mol Autism, 7; 27, 2016![]()
53) Woestelandt, L., Novo, A., Philippe, A., et al.: PDD-NOS, psychotic features and executive function deficits in a boy with proximal 22q11.2 microduplication: Evolution of the psychiatric symptom profile from childhood to adolescence. Eur J Med Genet, 61 (5); 280-283, 2018![]()
54) Yang, Y. M., Fedchyshyn, M. J., Grande, G., et al.: Septins regulate developmental switching from microdomain to nanodomain coupling of Ca (2+) influx to neurotransmitter release at a central synapse. Neuron, 67 (1); 100-115, 2010![]()
55) Yokoi, S., Ansai, S., Kinoshita, M., et al.: Mate-guarding behavior enhances male reproductive success via familiarization with mating partners in medaka fish. Front Zool, 13; 21, 2016![]()
56) Zweier, C., Sticht, H., Aydin-Yaylagül, I., et al.: Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet, 80 (3); 510-517, 2007![]()


